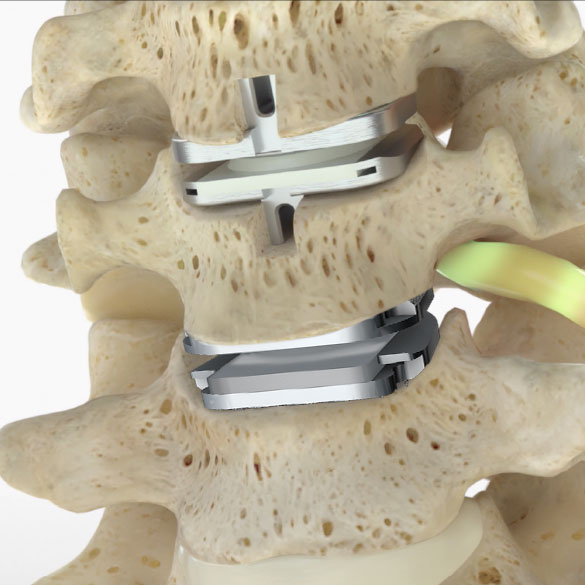
SMART™ clinical study
What is the
SMART™ Cervical Total Disc Replacement
Clinical Study?
The SMART Study is a nationwide clinical trial investigating the treatment of Two-Level Symptomatic Cervical Disc Disease (SCDD). The purpose of the trial is to evaluate the safety and effectiveness of two investigational devices, the prodisc® C SK and prodisc® C Vivo compared to the Mobi-C® Cervical Total Disc Replacement.